






Articulations trop souples !
Quand un apparent avantage peut devenir une maladie.



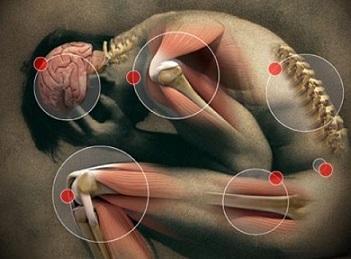
La souplesse des articulations a tendance à passer pour un avantage, du moins auprès de ceux qui avouent être « raides comme un piquet ». On admire celui qui parvient à mettre son pied derrière la tête, à faire le grand écart ou à placer les mains complètement à plat sur le sol. Et que dire des spectacles de contorsionnisme ! Cependant, quand la souplesse est extrême, la pathologie pourrait bien prendre le pas sur l’exploit ! C’est ce que nous a expliqué la Pre Valérie Gangji, cheffe du service rhumatologie et médecine physique de l’Hôpital Erasme et spécialiste du syndrome d’Ehlers-Danlos. « On parle de syndrome et non de maladie car il s’agit d’un ensemble de symptômes, précise-t-elle. Ce trouble du spectre de l’« hypermobilité » est aussi appelé « trouble de l’hyperlaxité ». Il s’agit au départ d’une souplesse extrême des articulations qui, lorsqu’elle devient très importante, porte alors le nom de syndrome d’Ehlers-Danlos hypermobile. On s’en rend généralement compte quand les patients commencent à développer régulièrement des luxations de l’épaule, des entorses, des douleurs articulaires, des dislocations… de façon spontanée ou lors de traumatismes mineurs. »
Ces symptômes s’accompagnent fréquemment d’autres troubles, comme une hyperextensibilité de la peau, des difficultés à cicatriser avec des cicatrices très fines dites en « papier à cigarette », l’apparition spontanée d’hématomes (propension à se faire « des bleus ») ou encore une tendance aux hémorragies. Cet ensemble de symptômes de l’hypermobilité constitue 95 % des cas. Pour les autres (5 % des cas), on a affaire à un syndrome d’Ehlers-Danlos de type vasculaire. « Dans ces cas heureusement rares, le patient présente une hyper-extensibilité de la paroi des vaisseaux, précise le Pr Gangji. Ceux-ci risquent des anévrismes (gonflements) et de se rompre. On peut aussi avoir une rupture d’organes tels que l’intestin ou l’utérus. Les conséquences dans ce cas peuvent être fatales. » Pour savoir si le patient présente une hyperlaxité préoccupante des doigts, des coudes, des poignets, des chevilles et des genoux, on peut le soumettre à un test (voir encadré) qui détermine les principaux critères de diagnostic du syndrome d’Ehlers-Danlos. Parmi ceux-ci, il y a, par exemple, la capacité à plier le poignet vers soi jusqu’à ce que le pouce touche l’avant-bras ou encore la facilité à déposer les mains à plat sur le sol, debout sans plier les genoux. Avec un score égal ou supérieur à cinq critères sur neuf, on peut penser être atteint du syndrome ED.

Mais d’autres critères peuvent constituer des signes de la maladie, comme l’apparition de vergetures, des problèmes gastro-intestinaux, des prolapsus (descente de vessie ou d’utérus) ou, chez la femme enceinte, une rupture prématurée et spontanée de la poche des eaux… Le syndrome d’ED affecte en effet la production de collagène (la protéine qui confère leur élasticité aux tissus conjonctifs et de soutien comme la peau, les tendons, les ligaments, la paroi des organes et celle des vaisseaux sanguins). Dans ce cas, les collagènes concernés sont anormaux et trop souples. Il s’agit d’une maladie génétique : elle peut apparaître spontanément, mais si un parent est porteur du gène, le risque de transmission à tous ses enfants est de 50 %. « En cas de suspicion de la forme vasculaire (rare) du syndrome ED, on peut faire une analyse génétique afin de déterminer la présence du gène, nous explique la Pre Gangji. Mais cette analyse n’est pas réalisée dans le syndrome d’ED hypermobile car il s’agit d’une atteinte polygénique dont on ne connaît pas actuellement les gènes anormaux. Il faut savoir enfin que le diagnostic ne peut être posé que si l’on n’a pas préalablement décelé une autre maladie rhumatismale inflammatoire. » Les principales complications que peut engendrer le syndrome d’Ehlers-Danlos sont des douleurs chroniques qui peuvent induire de la fatigue, un mauvais sommeil, de la peine à accomplir des exercices physiques, de l’arthrose, des symptômes cardiovasculaires, une ostéoporose précoce…

Pas de traitement précis
Cette maladie rare (la prévalence est d’environ une personne sur 5.000) semble affecter davantage les femmes que les hommes. Ses débuts sont difficiles à observer chez les enfants, les plus jeunes étant naturellement plus souples que leurs aînés et ne se plaignant pas. Certains signes d’hyperlaxité articulaire peuvent néanmoins déjà alerter, par exemple chez les enfants qui, couchés sur le dos, ont les pieds qui retombent naturellement sur les côtés extérieurs. Tout cela est-il grave ? Oui et non. Si elle ne raccourcit pas la vie, la forme non vasculaire du syndrome d’Ehlers-Danlos peut cependant diminuer considérablement la qualité de celle-ci. On traitera donc ce syndrome, non dans l’espoir de le guérir, mais dans le but d’en atténuer le plus possible les symptômes afin d’éviter les complications. Les antalgiques, les anti-inflammatoires, la kinésithérapie et l’exercice physique font donc partie de l’arsenal thérapeutique. Le sport n’est pas contre-indiqué, à condition d’éviter les traumatismes. Surveiller son poids est toujours utile pour éviter d’aggraver les douleurs articulaires et musculaires. Dans certains cas, des vêtements de contention en néoprène peuvent également soulager, qui agissent à la manière d’un exoquelette.

Un travail injustement oublié
On doit la première description de la maladie au dermatologue russe Alexandre Nicolaiev Tschernogobouw qui, en 1891, présenta deux patients souffrant d’une peau fragile et d’hypermobilité articulaire à la Société de dermatologie et de vénérologie de Moscou. Ironie du sort, son travail fut injustement oublié car le syndrome doit finalement son nom à deux autres dermatologues, l’un danois, le Dr Edvard Lauritz Ehlers, et l’autre français, le Dr Henri-Alexandre Danlos, lesquels ont respectivement décrit la maladie en 1900 et en 1908.
(Infos Gesed – Groupe d’entraide des syndromes d’Ehlers-Danlos)
Critères de diagnostic du syndrome d’Ehlers-Danlos hypermobile
A. Signes cliniques (il en faut au moins 5 sur 12)
– Peau douce et veloutée
– Extensibilité cutanée modérée
– Stries atrophiques ou rougeâtres, vergetures
– Papules piezogéniques au talon
– Hernie récurrente ou multiple (inguinale, crurale, hiatale, ombilicale)
– Cicatrices atrophiques (au moins deux sites)
– Prolapsus du plancher pelvien, rectal ou utérin sans antécédents majeurs
– Dentition irrégulière ET palais haut ou étroit
– Arachnodactylie (deux index-poignet ou deux pouces)
– Rapport envergure sur taille au moins 1,05
– Prolapsus mitral
– Dilatation de la racine de l’aorte avec Z-score supérieur à +2
B. Histoire familiale du syndrome ED
C. Atteintes musculo-squelettiques (au moins 1 point sur les 3)
1. Douleurs musculo-squelettiques d’au moins 2 membres, tous les jours et depuis plus de 3 mois.
2. Douleurs diffuses depuis plus de 3 mois.
3. Dislocation ou instabilité articulaire, sans traumatisme (au moins 1 point sur 2) :
– Au moins trois dislocations de la même articulation OU au moins deux dislocations pour deux articulations différentes.
– Instabilité d’au moins deux sites sans traumatisme auparavant.
Selon les critères de New York 2017.











